生物催化剂具有高效率、高选择性和可进化等优点,利用酶实现不对称催化合成具有重要意义。自然界中的酶大都已经进化成催化特定反应的生物催化剂,而利用酶实现非自然反应具有一定的挑战性。近年来,随着光化学的迅速发展,光化学中温和的反应条件和高反应性自由基中间体为酶催化反应类型的拓展提供了更多的可能性。目前,光酶催化非天然转化主要局限于单分子反应(例如:Hyster, Nature 2016, 540, 414-417; Zhao & Hartwig, Nature 2018, 560, 355-359等)。直到最近,伊利诺伊大学厄巴纳-香槟分校(UIUC)赵惠民团队(Nature 2020, 584, 69-74)和Hyster团队(J. Am. Chem. Soc. 2021, 143, 97-102)才先后实现了分子间的不对称合成。开发新的策略,实现更多非天然的分子间不对称合成是生物合成领域的研究热点之一。
近日,UIUC赵惠民教授、厦门大学王斌举教授和中科院深圳先进院周佳海研究员等人利用“化学模拟”策略,发展了一例新颖的光酶催化分子间不对称自由基共轭加成反应。以N-羟基邻苯二甲酰亚胺酯为自由基前体,利用其与酮还原酶活性位点中的NADPH形成的电子供体-受体(EDA)复合物,在光照下产生自由基。随后自由基对α,α-二取代末端烯烃进行加成得到前手性自由基,最后通过立体选择性的氢原子转移(HAT)构建α-羰基手性立体中心(图1)。

图1. NADPH依赖的酮还原酶催化的自由基反应。图片来源:Nat. Catal
首先,作者对商品化的酮还原酶进行了筛选,发现与来自开菲尔乳杆菌(Lactobacillus kefir)的野生型KRED相比,含有三个突变位点(F147L、L153Q、Y190P)的P2-D12能够以20%的产率,91:9的er值得到目标产物。为了探究底物与P2-D12之间的相互作用,作者对P2-D12-2a复合物的晶体结构进行了解析,分辨率为1.60 Å(图2)。从对结构的分析可以得知,底物2a结合在NADPH附近的疏水口袋中,2a上的乙烯基与NADPH之间形成π-π堆积相互作用,羰基与Ser143及Glu145之间形成氢键。190-210残基构成的柔性Loop灵活程度较高,控制着活性口袋中底物的进入和产物的释放。
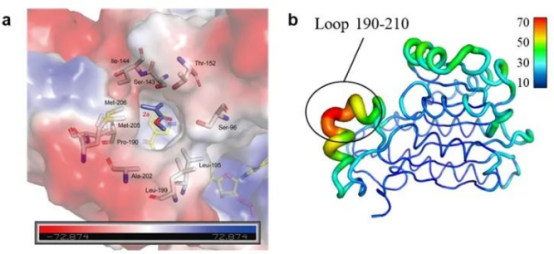
图2. P2-D12-2a复合物晶体结构。图片来源:Nat. Catal
作者进一步对酮还原酶进行了半理性迭代位点特异性突变,选取了活性位点附近的10个氨基酸残基进行突变。考虑到空间位阻的影响,作者只选取了以下氨基酸作为替换后的氨基酸:丙氨酸(A)或甘氨酸(G),亮氨酸(L)或异亮氨酸(I)以及苯丙氨酸(F)。在第一轮突变中,P2-D12_M206F (K1) 能够以9%的产率,93.5:6.5的er值得到产物。基于以上结果,作者以K1为母本,选取M205、A202、L199、P190 和 I144残基 用于第二轮突变,得到了三个具有改进对映选择性的KRED突变体:P2-D12_M206F/L199A (K2)、M206F/L199M206F/M205A,最后再经过一轮位点突变得到的P2-D12_M206F/L199A/M205F (K3) 能够催化该反应以 13% 的产率和 97:3 的er值生成产物。紧接着,作者通过对温度、NADPH再生系统、反应介质以及反应物1a/2a比例等条件的优化,用K3裂解液催化反应能够以79%的产率和 95.5:4.5的er值得到3a。

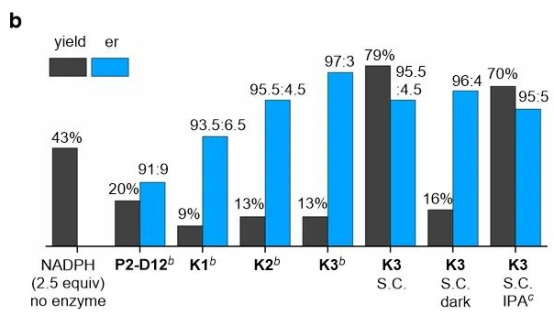

图3. 酶的进化。图片来源:Nat. Catal.
接着,作者进行了底物扩展(图4)。一系列苯乙酸衍生的NHPI酯都可以得到α-手性酯3a-3k,er值最高达96:4。苯环上取代基的位置对该反应影响不大(3c、3d、3h-k),但取代基的电子性质对反应结果有一定影响。例如,对三氟甲基取代1g为底物的反应进行得很慢。除苄基外,不同种类的叔碳自由基(3n-p)、仲碳自由基(3q)和伯碳自由基(3r-t)都能够进行对映选择性加成。此外,具有一个吸电子基团的α,α-二取代烯烃,包括甲基丙烯酰胺(3u)、吡啶基-2-烯烃(3v)和其它α,β-不饱和酯(3w-y)作为自由基受体时,该反应都能够顺利进行。

图4. 底物拓展。图片来源:Nat. Catal.
为了进行深入的机制研究,作者解析了K3-2a及K3-NHPI的复合物晶体结构,分辨率分别为1.56 Å和1.73 Å。此外,作者通过多尺度模拟进行了理论验证,包括经典的分子动力学(MD)模拟、QM/MM(量子力学/分子力学)MD模拟和QM模型计算。含时密度泛函理论(TDDFT)计算表明,1a的酰亚胺部分和NADPH之间通过π-π堆积形成的EDA复合物是唯一能够在可见光范围有吸收的合适构象。进一步的MD模拟确定了KRED活性位点中1a的稳定结合构象(图5b)。1a和NADPH之间的π-π堆积、1a和S143之间的氢键作用以及Y156的空间位阻都有助于1a与酶的结合。NHPI与K3复合物的晶体结构(图5h)显示出与EDA复合物的模拟构象相似的结合模式(图5b)。通过自由基验证实验观察到了5-己烯基的自由基环化反应,这与自由基的生成是一致的。
在2a的CH3-out 结合构象中(图5d),羰基与Ser143 结合,这与2a与K3复合物的晶体结构一致(图5i)。接下来,苄基自由基与烯烃2a的共轭加成得到前手性自由基,其势垒为7.2 kcal/mol(图5d)。从相邻的NADPH•+到前手性自由基的Si面HAT生成具有11.7 kcal/mol势垒的产物 (R)-3a(图5e)。作者通过氘标记实验进一步证实了这一观察结果,证明H原子完全来自原位产生的NADPH(图5f)。作者还研究了2a(CH3-in)的替代结合构象中的C-C键形成,反应将形成 (S)-3a。与 2a(CH3-in) 形成 (S)-3a(ΔG≠ = 9.7 kcal/mol,图5g)的势垒比形成 (R)-3a的势垒高2.5 kcal/mol(图5d)。所有模拟结果都与实验结果吻合,即反应主要形成 (R)-3a 产物。
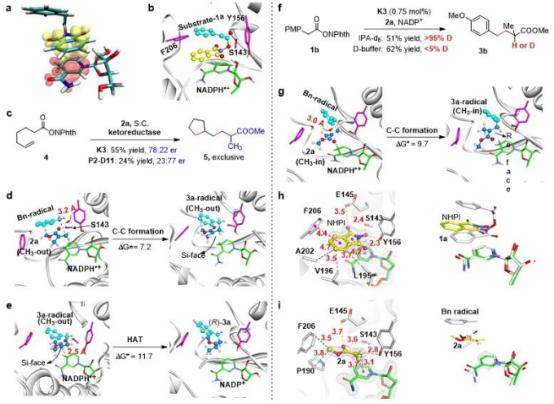
图5. 机理探究。图片来源:Nat. Catal
总体而言,本文报道了一例新颖的可见光诱导的生物催化自由基共轭加成反应,实现了自然界中没有的全新生物催化转化,扩展了酶的合成功能,对现有化学方法起到很好的互补作用。本文对机理的深入研究有助于开发更多非自然的生物转化,从而实现更多传统化学催化难以实现的转化。
文章来源:Photoinduced chemomimetic biocatalysis for enantioselective intermolecular radical conjugate additionXiaoqiang Huang, Jianqiang Feng, Jiawen Cui, Guangde Jiang, Wesley Harrison, Xin Zhang, Jiahai Zhou, Binju Wang, Huimin ZhaoNat. Catal., 2022, DOI: 10.1038/s41929-022-00777-4.